MDS-Symposium 2019
08. bis 11. Mai, Kopenhagen
- MDS-Symposium 2019: Time to get personal?
Dr. med. Stefani Barbara Parmentier, Rems-Murr-Klinikum, Winnenden

Auf dem 15. internationalen MDS-Symposium in Kopenhagen fand, wie erhofft, wieder ein reger Wissensaustausch zur Thematik der myelodysplastischen Syndrome (MDS) statt. Das Hauptaugenmerk lag in diesem Jahr auf der Biologie und der Pathophysiologie der MDS. Zahlreiche renommierte Spezialisten präsentierten dazu Vorträge, die es den Zuhörern ermöglichten, tiefere Einblicke in die Entstehung der MDS zu gewinnen. Mit diesem erweiterten Basiswissen lassen sich die neuen, äußerst interessanten Therapieansätze einfacher einordnen und besser verstehen, wobei die tatsächliche Umsetzung im ärztlichen Alltag noch vor uns liegt. Dennoch kann mit großer Hoffnung in die Zukunft geblickt werden. Seien Sie gespannt auf die neuen Erkenntnisse, die Sie in diesem Expertenbericht kompakt und übersichtlich zusammengefasst finden.
Liebe Kolleginnen und Kollegen,
das diesjährige MDS-Symposium fand vom 8. bis 11. Mai in Kopenhagen statt. Dabei präsentierten renommierte Wissenschaftler wieder die aktuellsten Erkenntnisse und Entwicklungen auf dem Gebiet der myelodysplastischen Syndrome (MDS). Ich freue mich sehr, Ihnen diese hier präsentieren zu dürfen.
Der Blick der Forscher war in diesem Jahr vor allem auf die zugrundeliegenden biologischen und pathophysiologischen Prozesse bei den MDS gerichtet. Das Verständnis dieser essenziellen Grundlagen ermöglichte es den Besuchern auch, die zahlreichen sehr interessanten neuen Therapieansätze besser nachzuvollziehen, die – insbesondere im Bereich der Immunologie – erarbeitet worden sind und nun auf dem MDS-Symposium ebenfalls vorgestellt wurden.
Ziel muss es sein, diese zukünftig in die Tat umzusetzen und personalisierte Therapieoptionen daraus zu entwickeln.
Welche praktischen Konsequenzen aus diesem breitgefächerten theoretischen Wissen entstehen, wird die Zukunft zeigen.
Ich wünsche Ihnen eine informative Lektüre, die Ihnen den einen oder anderen Denkanstoß für Ihren ärztlichen Alltag geben kann.
Mit kollegialen Grüßen
 Dr. med. Stefani Barbara Parmentier, Rems-Murr-Klinikum Winnenden
Dr. med. Stefani Barbara Parmentier, Rems-Murr-Klinikum WinnendenMDS-Symposium 2019: Time to get personal?
Dr. med. Stefani Barbara Parmentier, Rems-Murr-Klinikum, Winnenden
 Während beim 14. MDS-Symposium im Jahr 2017 die neue WHO-Klassifikation und neue therapeutische Optionen im Vordergrund standen, lag beim diesjährigen, 15. internationalen MDS-Symposium, der Fokus auf dem tiefergehenden Verständnis der Biologie der myelodysplastischen Syndrome (MDS). Hochkarätige Redner präsentierten sehr spannende Vorträge, bei denen es um die verbesserten Einblicke in die Pathophysiologie der MDS ging. Dabei wurde der Stellenwert immunologischer Prozesse betont. Auf dieser Basis bieten sich neue vielversprechende Therapieansätze, deren Einzug in den klinischen Alltag noch etwas Zeit beanspruchen wird. Diese neuen Behandlungsoptionen gilt es in der nahen Zukunft zu etablieren, um damit auch die personalisierten Therapien weiterzuentwickeln. Somit kann mit großer Spannung auf das nächste MDS-Symposium, das im Jahr 2021 stattfinden wird, geblickt werden.
Während beim 14. MDS-Symposium im Jahr 2017 die neue WHO-Klassifikation und neue therapeutische Optionen im Vordergrund standen, lag beim diesjährigen, 15. internationalen MDS-Symposium, der Fokus auf dem tiefergehenden Verständnis der Biologie der myelodysplastischen Syndrome (MDS). Hochkarätige Redner präsentierten sehr spannende Vorträge, bei denen es um die verbesserten Einblicke in die Pathophysiologie der MDS ging. Dabei wurde der Stellenwert immunologischer Prozesse betont. Auf dieser Basis bieten sich neue vielversprechende Therapieansätze, deren Einzug in den klinischen Alltag noch etwas Zeit beanspruchen wird. Diese neuen Behandlungsoptionen gilt es in der nahen Zukunft zu etablieren, um damit auch die personalisierten Therapien weiterzuentwickeln. Somit kann mit großer Spannung auf das nächste MDS-Symposium, das im Jahr 2021 stattfinden wird, geblickt werden.
Pathogenese des MDS
Für viele maligne Erkrankungen konnten bereits chronisch inflammatorische Prozesse als Ursache ihrer Pathogenese identifiziert werden. Bei den myelodysplastischen Syndromen (MDS) dagegen wurde lange Zeit deren potenzielle Rolle nicht sicher nachgewiesen. Das hat sich vor kurzem geändert. Mufti et al. berichteten schon 1986 in einer Arbeit über den Zusammenhang von immunologischen Veränderungen und dem Vorliegen eines MDS [1]. Die Ergebnisse einer weiteren Studie konnten zudem zeigen, dass das Vorliegen einer Autoimmunerkrankung mit einer Verbesserung des Gesamtüberlebens (OS) beim MDS zusammenhängt [2]. Dies scheint auf einem initial protektiven Effekt des adaptiven Immunsystems zu beruhen [3]. Erika Eksioglu [4] und Shahram Kordasti [5] griffen diese Vermutung nun aktuell auch in ihren Vorträgen auf. Dabei sprachen sie einer altersbedingten chronischen Inflammation eine entscheidende Rolle bei der möglichen Entwicklung eines MDS zu. Von beiden Rednern wurde das vermehrte Vorkommen von myeloiden Suppressorzellen (Myeloid-derived Suppressor Cells, MDSCs) erwähnt, aus denen unter anderem S100A9, ein DAMP-Protein (DAMP, Damage-associated molecular Pattern), freigesetzt wird. S100A9 führt zu einer Aktivierung des Inflammasoms NLRP3 (NOD[Nucleotide Oligomerization Domain]-like-Receptor-Protein 3), was wiederum den Zelltod, die Pyroptose, bedingt.
Kordasti et al. [6] konnten erstmalig einen erhöhten Anteil an TH17-Zellen (Interleukin-17 produzierende T-Helferzellen) sowie ein erhöhtes TH17-zu-Treg-Verhältnis (Treg, regulatorische T-Helferzelle) bei Niedrigrisiko-MDS nachweisen.
Während seines Vortrags in Kopenhagen stellte Shahram Kordasti nun eine neue Form von regulatorischen T-Zellen vor – die CD161-positiven Tregs [5]. Diese führen nach ihrer Stimulation zu einer vermehrten Freisetzung von Interleukin-10 (IL-10), welches über ein hohes immunsuppressives Potenzial verfügt. Es konnte gezeigt werden, dass bei Patienten mit einer geringen Anzahl CD161+-Tregs ein besseres Ansprechen auf Luspatercept erreicht wird. Multiple Faktoren, wie MDSCs, TGF-β (Transforming Growth Factor-β), PD-L1 (Programmed-Cell-Death-Ligand 1) und Tregs, beeinflussen also entscheidend die Toleranz und Immunität bei Patienten mit MDS.
Des Weiteren konnte bereits mehrfach gezeigt werden, dass das Mikroenvironment beziehungsweise die mesenchymale Stammzellnische eine Schlüsselrolle bei der Krankheitsentstehung und -progression der MDS spielt [7]. Marc Raaijmakers ging in seinem Vortrag auf diesen wesentlichen Pathomechanismus ein (Abb. 1) [8, 9]. Mesenchymale Nischenzellen induzieren genotoxischen Stress in hämatopoetischen Stammzellen (HSCs) beziehungsweise Vorläuferzellen (HSPCs). Dies ist prädiktiv für den Progress in eine akute myeloische Leukämie (AML) [7].
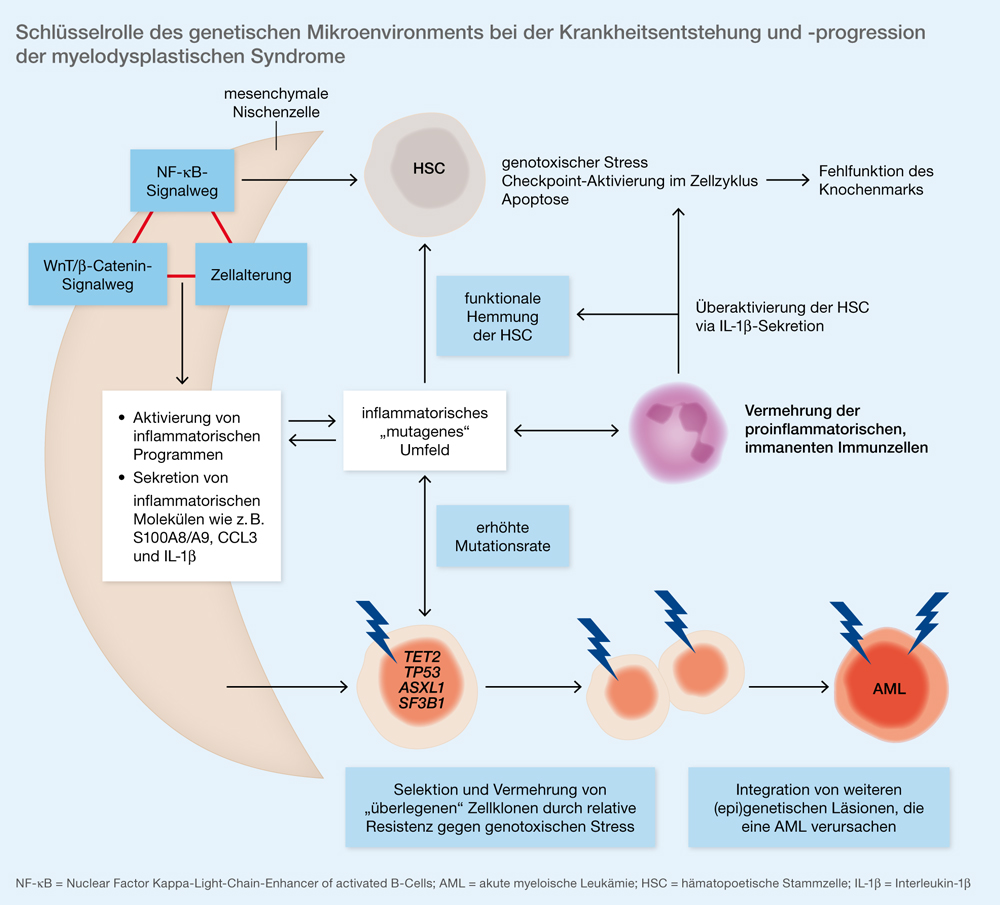
Die MDSCs bei Patienten mit MDS können einerseits direkt mittels Anti-CD33, andererseits indirekt durch die Einleitung der Differenzierung mithilfe von ATRA (All-trans-Retinsäure) oder die Hemmung von S100A9 und PD-1 (Programmed-Cell-Death-Protein 1)/PD-L1 (Programmed-Cell-Death-Ligand 1) gehemmt werden. S100A9 scheint allerdings kein sinnvolles Target zu sein, da es bei zunehmenden Hochrisiko-MDS herunterreguliert wird.
Marc Raaijmakers berichtete des Weiteren über den Einfluss veränderter Makrophagen bei den MDS [8]. Makrophagen, mesenchymale Stammzellen und tubulo-interstitielle Zellen der Niere spielen eine wichtige Rolle bei der Erythropoese. Raajimakers stellte zudem Daten von Daniel Pollyea [11] vor, die zeigen, dass Spliceosom-Mutationen (SRSF2, U2AF1, SF3B1) zu einer verstärkten Freisetzung von inflammatorischen Zytokinen (IL-6 und NF-κB [Nuclear Factor Kappa-Light-Chain-Enhancer of activated B-Cells]) aus den Makrophagen führen. Bei den veränderten Makrophagen führt eine Überexpression von S100A8 und S100A9 zur Anämie bei bestimmten Subtypen der MDS.
Rafael Bejar berichtete in seinem Vortrag, dass aberrante Splicing-Mutationen von Schlüsselgenen (ABCB7, SLC25A37, TMEM14C, PPOX) den Phänotyp „Ringsideroblasten“ zu vermitteln scheinen [12]. Man konnte zudem zeigen, dass Patienten mit SF3B1-mutiertem MDS seltener Co-Mutationen in Genen zeigten, die mit einer schlechteren Prognose assoziiert sind.
Darüber hinaus zeigen verschiedene Studien [13, 14, 15], dass der Verlust des TET2-Gens zu unkontrollierter Inflammation führt, die vor allem durch Interleukin(IL)-6 und IL-1β vermittelt wird. TET2-mutierte Zellen sind dabei resistent gegenüber inflammationsvermittelter Hemmung und Apoptose hämatopoetischer Stammzellen.
Fazit
- Multiple Faktoren, wie MDSCs, TGF-β, PD-L1 und Tregs, beeinflussen die Toleranz und Immunität bei den MDS.
- MDSCs sind bei MDS-Patienten erhöht und nehmen bei Patienten mit Hochrisiko-MDS deutlich zu.
- Verschiedene, epigenetische und Splicing-Mutationen bei MDS führen zu Veränderungen im angeborenen Immunsystem, insbesondere in Makrophagen, bei denen eine Überexpression von S100A8 und S100A9 zur Anämie bei bestimmten Subtypen von MDS beiträgt.
- Aberrante Splicing-Mutationen von Schlüsselgenen (ABCB7, SLC25A37, TMEM14C, PPOX) scheinen den Phänotyp „Ringsideroblasten“ zu vermitteln.
- MDSCs können bei MDS somit direkt über Anti-CD33, aber auch indirekt durch die Einleitung der Differenzierung mittels ATRA oder die Hemmung von S100A9 und PD-1/PD-L1 gehemmt werden. Allerdings wird S100A9 bei zunehmenden Hochrisiko-MDS herunterreguliert.
“Indem wir die Rolle des Immunsystems bei den MDS zunehmend besser verstehen, können hier zielgerichtete und hoffentlich vielversprechende Therapieansätze entwickelt werden.” Dr. med. Stefani Parmentier
Therapie der Niedrigrisiko-MDS
Mit zunehmendem Verständnis der Pathophysiologie der MDS wird auch die Heterogenität der Erkrankung immer deutlicher. Daher bemüht man sich unter anderem, zielgerichtete Therapien zu entwickeln. Es ist immer wieder zu betonen, dass heute zu einer adäquaten Diagnostik bei MDS auch die Bestimmung molekularer Marker gehört, welche wichtige prognostische und prädiktive Aussagen in Bezug auf das Ansprechen einer Therapie liefern können. Ein bekanntes Beispiel dafür ist Lenalidomid beim MDS mit 5q-Deletion (del(5q)).
Elli Papaemmanuil stellte einen genetischen Algorithmus für die Therapie der MDS vor [16]. Dabei stehen SF3B1-Mutationen, die mit einer eher guten Prognose (bei Niedrigrisiko-MDS ohne Blastenmutation) einhergehen, Mutationen gegenüber, die mit einer intermediären (z. B. TET2, DNMT3A, ZRSR2, IDH1) beziehungsweise schlechten Prognose (z. B. ASXL1, RUNX1, TP53, NRAS) assoziiert sind. Dieses Wissen wird in einen neuen Risikoscore, das IPSS-Rm (Revised-international-Prognostic-Scoring-System „molecular“), einfließen. Dabei werden die molekularen Marker berücksichtigt. Die Veröffentlichung wird für 2020 erwartet.
Uwe Platzbecker gab in seinem Vortrag [17] sowie in seiner Publikation [18] einen sehr schönen Überblick zu den aktuellen Therapiealgorithmen bei Niedrigrisiko- und Hochrisiko-MDS.
Liegen Niedrigrisiko-MDS vor, so steht die Verbesserung der Zytopenien sowie die Vermeidung deren Komplikationen im Fokus der Therapieentscheidungen. Obwohl erythropoesestimulierende Substanzen (ESAs) seit vielen Jahren eingesetzt werden, erfolgte erst vor Kurzem – basierend auf den Ergebnissen prospektiver Studien – die Zulassung von Epoetin-α (EPO-α) [19, 20]. Vor dem Einsatz von EPO-α sollte allerdings ein Erythropoetinspiegel < 200 IU/l vorliegen. Ein Ansprechen auf die Gabe von EPO-α ist in der Regel durchschnittlich innerhalb von 3 Monaten zu verzeichnen – mit einer medianen Wirkdauer von 15 bis 18 Monaten. Bei Patienten, die nicht auf ESAs ansprechen oder bei denen die Wirksamkeit im Verlauf abnimmt, kann die Hinzunahme des Granulozyten-Kolonie-stimulierenden Faktors (G-CSF, Granulocyte-Colony stimulating Factor) in Erwägung gezogen werden, was in immerhin 20% der Fälle ein Ansprechen bewirkte [21]. Dies scheint insbesondere bei Patienten mit MDS und Ringsideroblasten (MDS-RS), die im Vergleich zu Patienten ohne RS ein zeitlich verkürztes Ansprechen auf ESAs zeigen, eine Rolle zu spielen [22].
Eva Hellström-Lindberg beleuchtete in ihrem Vortrag die Bedeutung von SF3B1-Mutationen bei MDS [23]. Diese Mutationen spielen in den hämatopoetischen Stammzellen (HCSs) eine entscheidende Rolle. Wenn keine Hochrisikomutationen hinzukommen, ist hier eine geringe Progressionsrate zu verzeichnen. Die Patienten mit SF3B1-Mutationen weisen im Verlauf lediglich einen hohen Transfusionsbedarf auf. Die Gabe von EPO +/- G-CSF wirkt dabei über antiapoptotische Mechanismen [24]. Wenn SF3B1-dominierende Klone vorliegen, führt dies zu einem ESA-refraktären Verlauf.
Vielversprechend bei der Behandlung von Patienten mit MDS-RS ist der Einsatz von Luspatercept, das bereits vor 2 Jahren beim MDS-Symposium in Valencia vorgestellt wurde (siehe auch Bericht vom MDS-Symposium 2017 auf hematooncology.com.) Manja Wobus griff dieses Thema in ihrem aktuellen Vortrag auf [24]. Liganden der TGF-β-Superfamilie (TGF-β, Transforming Growth Factor-β) führen zu einer vermehrten Aktivität der SMAD2/3-Proteine, welche die Erythropoese hemmen. Luspatercept wiederum hemmt die Liganden (“ligand trap”) der TGF-β-Familie und somit auch die gesteigerte Aktivität von SMAD2/3, was zu einer Verbesserung der Erythropoese führt [24, 25]. Als Hemmer des Liganden GDF-11 (GDF-11, Growth Differentiation Factor 11) am Activin-Rezeptor-Typ-IIb (AcvR2B) führt Luspatercept schließlich zu einer Abnahme der Transfusionsfrequenz (Abb. 2). Die Ergebnisse einer Phase-II-Studie mit Luspatercept zeigten bei 63% der Patienten ein Ansprechen in Bezug auf die roten Blutzellen und 38% der Patienten erreichten eine Transfusionsunabhängigkeit [18, 25, 26, 27].

Uwe Platzbecker betonte zudem in seinem Vortrag [27], dass bei transfusionsabhängigen Patienten, insbesondere mit einer Lebenserwartung > 12 Monate oder bei einer geplanten Stammzelltransplantation, eine Eisenchelation in Betracht gezogen werden sollte (Abb. 3). In der TELESTO-Studie konnte so eine Risikoreduktion von 36,4% in Bezug auf das ereignisfreie Überleben (EFS) erreicht werden [28, 29].
Was neue Therapieoptionen bei MDS angeht, ging Uwe Platzbecker in seinem Vortrag unter anderem auf Roxadustat (FG-4592), einen Hypoxie induzierenden Faktor-Prolylhydroxylase-Inhibitor (HIF-PHI), ein [17]. Diese Substanz wird gerade in einer Phase-III-Studie zur Behandlung von Patienten mit Anämie bei Niedrigrisiko-MDS und niedriger Transfusionslast für Erythrozytenkonzentrate (EKs) untersucht (NCT03263091). Roxadustat bewirkt über einen Anstieg des endogenen Erythropoetinspiegels sowie über eine verbesserte Eisenregulierung mittels Modulierung des Hepcidinspiegels, eine Verbesserung der Erythropoese.
Telomere und Telomerase sind wichtig für die Aufrechterhaltung einer normalen Hämatopoese. Es konnte gezeigt werden, dass bei Patienten mit MDS deutlich kürzere Telomerlängen und eine erhöhte Telomeraseaktivität vorliegen [30]. Vorläufige Ergebnisse einer Phase-II/III-Studie mit dem Telomerasehemmer Imetelstat konnten zeigen, dass 37% der Patienten mit Transfusionsabhängigkeit und ESA-Versagen eine Transfusionsunabhängigkeit erreichen konnten. Daneben ließ sich ein Abfall der Mutationslast verzeichnen [17, 31, 32].
Bei Patienten mit Niedrigrisiko-MDS und gleichzeitiger Thrombozytopenie konnte unter Therapie mit Thrombopoetin-Rezeptor-Agonisten (TPO-RAs) ein Ansprechen bis nahezu 50% verzeichnet werden [33]. Aktuell laufen allerdings noch weitere Studien (NCT02335268), sodass die Ergebnisse hierzu noch abgewartet werden müssen [18].
Fazit
- ESAs (EPO-α) stehen nun für die Therapie des Niedrigrisiko-MDS zur Verfügung.
- Bei hohem Transfusionsbedarf sollte bei ansonsten guter Lebenserwartung und fehlenden Kontraindikationen, vor allem vor einer allogenen Stammzelltransplantation (SCT), mit einer Eisenchelation begonnen werden.
- Aktuell noch laufende Studien zu Roxadustat und Imetelstat scheinen bei relativ guter Verträglichkeit in Bezug auf eine Verbesserung der Transfusionsfrequenz effektiv zu sein.
- Die Daten zu Luspatercept in der Therapie der MDS-RS sind sehr vielversprechend, sodass eine Zulassung für 2020 erwartet wird.
“Mit Roxadustat, Imetelstat und Luspatercept können wir auf vielversprechende Substanzen in der Therapie des Niedrigrisiko-MDS hoffen.” Dr. med. Stefani Parmentier
Therapie der Hoch-Risiko MDS und CMML
Bei Hochrisiko-MDS ist die Prognose sehr schlecht, insbesondere nach dem Versagen einer Therapie mit Azacitidin (AZA) oder wenn für die betroffenen Patienten die Option einer allogenen SCT nicht zur Verfügung steht. Guillermo Garcia-Manero berichtete von aktuellen Studien mit Guadecitabin, einer hypomethylierenden Substanz (HMA) der Zweitlinie. Deren Gabe konnte den Untersuchungen zufolge auch nach einem AZA-Versagen noch ein Ansprechen erreichen. Allerdings handelt es sich hierbei noch um vorläufige Ergebnisse, da diese Studien noch nicht beendet sind. Die präsentierten Daten scheinen bei der chronisch myelomonozytären Leukämie (CMML) sehr vielversprechend [34]. Therapienative Patienten boten hierbei Komplettremissionsraten (CR) von 22% mit einem medianen OS von 23,4 Monaten. Bei Patienten, die bereits vortherapiert waren, ließen sich immerhin noch CRs (mit noch positiver minimaler residueller Resterkrankung, MRD) von 32% und ein medianes OS von 11,7 Monaten erreichen. Dabei ließ sich bei Patienten mit einer Therapiedauer von mindestens 4 bis 6 Zyklen ein signifikant längeres Überleben zeigen als für Patienten, die weniger Zyklen erhalten haben. Aktuell läuft noch eine Phase-III-Studie für rezidivierte/refraktäre MDS/CMML (ASTRAL-3-Studie, NCT02907359).
Alan Shih konnte in Mausmodellen zeigen, dass die Therapie mit dem MEK-Inhibitor Binimetinib beim Vorliegen einer CMML mit TET2/NRAS-Doppelmutation und zusätzlicher AZA-Resistenz ein gutes Ansprechen erreicht [35]. Um jedoch eine abschließende Aussage darüber treffen zu können, müssen noch weitere Studienergebnisse abgewartet werden.
Die Kombination von HMAs und Immuncheckpointinhibitoren (ICPIs), wie zum Beispiel dem Programmed-Cell-Death-Ligand-1(PD-L1)-Inhibitor (Anti-PD-L1), ist sehr effektiv und zeigt ein gutes molekulares Ansprechen beim Vorliegen einer TP53-Mutation, allerdings um den Preis einer hohen Toxizität. Immuncheckpoints (ICs) bewirken ein Herabregulieren der T-Zell-Aktivierung oder der T-Zell-Effektorfunktion. Tumorzellen, die sich dieser ICs bedienen können, sind fähig, der Erkennung durch das Immunsystem zu entkommen (Immunevasion). Guillermo Garcia-Manero erläuterte in seinem Vortrag weitere Ergebnisse dazu [36]. Während die Studie mit Pembrolizumab vor allem aufgrund erhöhter Toxizitäten abgebrochen werden musste, wurden hohe Ansprechraten für Nivolumab+AZA (40% CR, 70% ORR) beschrieben. Das mediane OS wurde bei Ipilimumab+AZA noch nicht erreicht. Ipilimumab zeigt auch ein Ansprechen als Einzelsubstanz nach dem Versagen von HMAs. Bei einer immunvermittelten Toxizität sollte frühzeitig die Gabe von Prednisolon erfolgen.
Enttäuschend waren die aktuellen Daten zur Therapie mit AZA in Kombination mit Lenalidomid, Valproat oder Idarubicin im Vergleich zur AZA-Monotherapie. Wie Lionel Ades berichten konnte, ergab sich kein Unterschied in Bezug auf das Ansprechen, das PFS oder das Gesamtüberleben (OS) [37].
Interessant waren Aussagen von Linn Gillberg [38] und Peter Jones [39] zum Zusammenhang zwischen MDS und dem Vitamin-C-Spiegel im Plasma. Bei MDS-Patienten scheint dieser regelhaft erniedrigt zu sein. Es sieht so aus, als führe sein Ausgleich über epigenetische Veränderungen zu einer verbesserten Wirkung von AZA.
Ebenfalls vielversprechend ist die Kombination von Venetoclax mit AZA, womit schon hohe Ansprechraten verzeichnet werden konnten. Die Studien hierzu laufen allerdings noch (NCT02966782).
Wie von Uwe Platzbecker ausgeführt, sollte vor der Einleitung einer Therapie bei Hochrisiko-MDS initial evaluiert werden, ob die Patienten für eine allogene SCT geeignet sind. Bis zur Transplantation sollte dann eine Eisenchelation erfolgen. Bei einem Progress in eine AML kann eventuell eine der neu zugelassenen Substanzen zur zielgerichteten Therapie (IDH[Isocitratdehydrogenase]- oder FLT3[Fms-like Tyrosine Kinase 3]-Inhibitoren) eingesetzt werden. Generell sollte stets geprüft werden, ob die Patienten an einer laufenden Studie teilnehmen können (Abb. 4).
Fazit
- Der Einsatz von HMAs, wie Guadecitabin, in der Zweitlinie scheint ein Ansprechen nach AZA-Versagen zu erreichen.
- Die Kombination von HMAs mit ICPIs (z. B. Anti-PD-L1) ist sehr effektiv und zeigt ein gutes molekulares Ansprechen bei TP53-Mutationen, allerdings um den Preis einer hohen Toxizität.
- AZA in Kombination mit Lenalidomid, Valproat oder Idarubicin ergab keinen Unterschied in Bezug auf das Ansprechen, das PFS oder das OS.
- Der Ausgleich niedriger Vitamin-C-Spiegel im Plasma führt über epigenetische Veränderungen zu einer verbesserten Wirkung von AZA.
- Venetoclax in Kombination mit AZA scheint zu hohen Ansprechraten zu führen. Die Studien hierzu laufen allerdings noch.
- Der MEK-Inhibitor Binimetinib zeigt beim Vorliegen einer CMML mit einer TET2/NRAS-Doppelmutation und einer zusätzlichen AZA-Resistenz ein Ansprechen im Mausmodell.
“In wenigen Jahren werden wir in der Lage sein, Niedrigrisiko- und Hochrisikopatienten effektive Therapieoptionen über AZA hinaus anbieten zu können. Damit erreichen wir zunehmend die Ebene einer zielgerichteten, personalisierten Medizin.“ Dr. med. Stefani Parmentier
Zusammenfassung
Beim diesjährigen MDS-Symposium in Kopenhagen lag der Schwerpunkt auf der Pathophysiologie und der Biologie der MDS. Dazu wurden äußerst gelungene Vorträge von hochrangigen Rednern gehalten. Dabei wurden zahlreiche neue interessante Therapieansätze aufgezeigt, bis zu deren Umsetzung es im klinischen Alltag aber noch etwas Geduld bedarf. Wir benötigen mehr Therapien. Die Erkenntnisse über das molekulargenetische Muster allein reichen nicht aus. Aber wir sollten die zunehmend zugänglichen Informationen über Molekular- und Zytogenetik dazu nutzen, auch hier weitere „personalisierte“ beziehungsweise zielgerichtete Therapien zu entwickeln. Hierzu gibt es sehr gute Ansätze – unter anderem mit neuen Substanzen wie Roxadustat, Imetelstat, Luspatercept und Immuncheckpointinhibitoren. Vielversprechend ist auch der Einsatz von Venetoclax.
Quellen
- Mufti GJ et al. Immunological abnormalities in myelodysplastic syndromes. I. Serum immunoglobulins and autoantibodies. Br J Haematol 1986; 63: 143-147.
- Komrokji RS et al. Autoimmune diseases and myelodysplastic syndromes. Am J Hematol 2016; 91: E280-283.
- Aggarwal S et al. Role of immune responses in the pathogenesis of low-risk MDS and high-risk MDS: implications for immunotherapy. Br J Haematol 2011; 153: 568-581.
- Eksioglu E. The S100A9/FTO axis induces formation of RNA: DNA hybrids damps and the dysregulation of normal splicing patterns in MDS. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Kordasti S. Immune dysregulation in MDS; an organised mess? Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Kordasti SY et al. IL-17-producing CD4(+) T cells, pro-inflammatory cytokines and apoptosis are increased in low risk myelodysplastic syndrome. Br J Haematol 2009; 145: 64-72.
- Zambetti NA et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-leukemia. Cell Stem Cell 2016; 19: 613-627.
- Raaijmakers M. The niche: the hen or the egg. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Raaijmakers M. The significance of the microenvironment in ineffective erythropoiesis. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Pronk E et al. The mesenchymal niche in MDS. Blood 2019; 133: 1031-1038.
- Pollyea DA et al. Myelodysplastic syndrome-associated spliceosome gene mutations enhance innate immune signaling. Haematologica 2019.
- Bejar R. Genetic changes underlying ineffective erythropoiesis. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Cull AH et al. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol 2017; 55: 56-70 e13.
- Zhang Q et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015; 525: 389-393.
- Cai Z et al. Inhibition of Inflammatory Signaling in Tet2 Mutant Preleukemic Cells Mitigates Stress-Induced Abnormalities and Clonal Hematopoiesis. Cell Stem Cell 2018; 23: 833-849.e835.
- Papaemmanuil E. A genetic algorithm for treatment. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Platzbecker U. Low risk disease-what`s new? Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Platzbecker U. Treatment of MDS. Blood 2019; 133: 1096-1107.
- Fenaux P et al. A phase 3 randomized, placebo-controlled study assessing the efficacy and safety of epoetin-alpha in anemic patients with low-risk MDS. Leukemia 2018; 32: 2648-2658.
- Platzbecker U et al. A phase 3 randomized placebo-controlled trial of darbepoetin alfa in patients with anemia and lower-risk myelodysplastic syndromes. Leukemia 2017; 31: 1944-1950.
- Kelaidi C et al. Long-term outcome of anemic lower-risk myelodysplastic syndromes without 5q deletion refractory to or relapsing after erythropoiesis-stimulating agents. Leukemia 2013; 27: 1283-1290.
- Park S et al. Outcome of Lower-Risk Patients With Myelodysplastic Syndromes Without 5q Deletion After Failure of Erythropoiesis-Stimulating Agents. J Clin Oncol 2017; 35: 1591-1597.
- Hellström-Lindberg E. MDS-RS: refractory anemia with ring sideroblasts. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Wobus M. The stromal support of hematopoiesis in MDS is improved by Luspatercept. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Platzbecker U et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol 2017; 18: 1338-1347.
- Fenaux P et al. Luspatercept for the treatment of anemia in myelodysplastic syndromes and primary myelofibrosis. Blood 2019; 133: 790-794.
- Platzbecker U. TGF-ß superfamily/SMAD signalling: a pathway to ineffective erythropoiesis. Oral presentation at the 15th Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Angelucci E et al. Safety and Efficacy, Including Event-Free Survival, of Deferasirox Versus Placebo in Iron-Overloaded Patients with Low- and Int-1-Risk Myelodysplastic Syndromes (MDS): Outcomes from the Randomized, Double-Blind Telesto Study. Blood 2018; 132: 234-234.
- Wermke M et al. Enhanced labile plasma iron and outcome in acute myeloid leukaemia and myelodysplastic syndrome after allogeneic haemopoietic cell transplantation (ALLIVE): a prospective, multicentre, observational trial. Lancet Haematol 2018; 5: e201-e210.
- Park HS et al. Dysregulation of Telomere Lengths and Telomerase Activity in Myelodysplastic Syndrome. Ann Lab Med 2017; 37: 195-203.
- Steensma DP et al. Imetelstat Treatment Leads to Durable Transfusion Independence (TI) in RBC Transfusion-Dependent (TD), Non-Del(5q) Lower Risk MDS Relapsed/Refractory to Erythropoiesis-Stimulating Agent (ESA) Who Are Lenalidomide (LEN) and HMA Naive. Blood 2018; 132: 463-463.
- Fenaux P et al. Efficacy and Safety of Imetelstat in RBC Transfusion-Dependent (TD) IPSS Low/Int-1 MDS Relapsed/Refractory to Erythropoiesis-Stimulating Agents (ESA) (IMerge). Blood 2017; 130: 4256-4256.
- Oliva EN et al. Eltrombopag versus placebo for low-risk myelodysplastic syndromes with thrombocytopenia (EQoL-MDS): phase 1 results of a single-blind, randomised, controlled, phase 2 superiority trial. Lancet Haematol 2017; 4: e127-e136.
- Garcia-Manero G. Long-term survival results and prognostic factors of higher risk myelodysplastic syndromes (MDS)/chronic myelomonocytic leukemia (CMML) treated with Guadecitabine. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Shih A. Preclinical models of CMML (TET2/NRAS) and sensitivity to MAPK inhibitor. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Garcia-Manero G. Combining Hypomethylation drugs and immune check point inhibitors. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Ades L. A randomized phase II study of Azacitidine alone or with Lenalidomide (LEN), Valproic Acid (VPA) or Idarubicin (IDA) in higher-risk MDS. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Gillberg L. Oral Vitamin C supplementation to myeloid cancer patients on Azacitidine treatment: normalization of plasma Vitamin C induces epigenetic changes. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Jones P. Epigenetic therapy and the immune system. Oral presentation at the 15th International Symposium on Myelodysplastic Syndromes, Copenhagen, May 2019.
- Bildnachweis: „Copenhagen”: © Andreas Gradin/Adobe Stock; "The entry gate of Tivoli Gardens": © lermont51/Adobe Stock