MDS-Forum 2016
22. bis 23. April, München
 MDS/AML 2016: Aktuelles Wissen in die Praxis überführt (Dr. Raoul Tibes, Würzburg)
MDS/AML 2016: Aktuelles Wissen in die Praxis überführt (Dr. Raoul Tibes, Würzburg)
Beim diesjährigen MDS-Forum, das vom 22. bis 23. April 2016 in München stattfand, gaben nationale und internationale Referenten einen umfassenden Überblick über den aktuellen Wissensstand zu MDS und AML – spannende Diskussionen zu offenen klinischen und molekularen Fragestellungen inklusive.
Liebe Kolleginnen und Kollegen,
der Titel des diesjährigen MDS-Forums, das am 22. und 23. April 2016 in München abgehalten wurde, war Programm: „MDS und AML from Bench to Bedside: Neues Wissen schneller in die Anwendung bringen“. Die Veranstaltung hatte sich zum Ziel gesetzt, neue Erkenntnisse zu Pathophysiologie und molekularen Mechanismen bei MDS und AML möglichst rasch in klinisch anwendbare diagnostische, prognostische, prädiktive und therapeutische Ansätze zu überführen.
Dieser Bericht gibt Ihnen einen Überblick über die diskutierten Themen und bietet Ihnen Anregungen für die tägliche Arbeit am Patient. Ich wünsche Ihnen eine interessante Lektüre und freue mich über Ihr Interesse.
Mit kollegialen Grüßen
Ihr Raoul Tibes
 Dr. med. Raoul Tibes, Universitätsklinikum Würzburg
Dr. med. Raoul Tibes, Universitätsklinikum WürzburgMDS/AML 2016: Aktuelles Wissen in die Praxis überführt
Dr. med. Raoul Tibes, Universitätsklinikum Würzburg
 Das Programm des diesjährigen MDS-Forums war hervorragend zusammengestellt. Nationale und internationale Referenten gaben einen umfassenden Überblick über das gesamte Gebiet bei MDS und AML – sowohl mit den wesentlichen Aspekten zum aktuellen Wissensstand als auch zu offenen klinischen und molekularen Fragestellungen. Ebenfalls sehr interessant war die Darstellung seltener myeloischer Erkrankungen. Da viele der Sessions Zusammenfassungen des aktuellen Standes von Forschung und Therapie waren und die Vorträge auf eine Synthese des momentanen Wissens abzielten, werden in diesem Kongressbericht ebenfalls Zusammenfassungen der jeweiligen Themenbereiche nach den einzelnen Sessions vorgenommen und ein breiter aktueller Überblick über alle wichtigen Aspekte bei MDS und AML dargestellt.
Das Programm des diesjährigen MDS-Forums war hervorragend zusammengestellt. Nationale und internationale Referenten gaben einen umfassenden Überblick über das gesamte Gebiet bei MDS und AML – sowohl mit den wesentlichen Aspekten zum aktuellen Wissensstand als auch zu offenen klinischen und molekularen Fragestellungen. Ebenfalls sehr interessant war die Darstellung seltener myeloischer Erkrankungen. Da viele der Sessions Zusammenfassungen des aktuellen Standes von Forschung und Therapie waren und die Vorträge auf eine Synthese des momentanen Wissens abzielten, werden in diesem Kongressbericht ebenfalls Zusammenfassungen der jeweiligen Themenbereiche nach den einzelnen Sessions vorgenommen und ein breiter aktueller Überblick über alle wichtigen Aspekte bei MDS und AML dargestellt.
Session 1: Pathophysiologie bei MDS und AML [1,2,3]
Im Hauptvortrag über die Pathophysiologie bei MDS und AML gab der international anerkannte Experte Rafael Bejar einen Überblick über die momentane Anwendung und Bedeutung von Mutationen und genetischen Veränderungen für die Prognose und das Therapieansprechen bei MDS. Mutationen beeinflussen oft negativ die Entwicklung hämatopoetischer Stamm- und Progenitorzellen und können eine kontinuierliche und regelrechte Hämatopoese verhindern. In diesem Zusammenhang ist wichtig, dass bis zu 2/3 der MDS-Patienten ihrer Krankheit durch Zytopenien erliegen und etwa 1/3 der Patienten nach der Transformation zu einer AML. Je nach Publikation sind es zwischen 20 und 40 charakteristische Mutationen, die wiederkehrend bei MDS beobachtet werden [4]. Neben der Forschung zur biologischen Rolle von Mutationen bei MDS zogen sich die folgenden klinischen Fragestellungen wie ein roter Faden durch den Kongress:
- Können Mutationen zur Diagnose herangezogen werden?
- Wie beeinflussen Mutationen die (natürliche) Prognose bei MDS und AML zusätzlich zu den bereits etablierten Risikofaktoren wie zytogenetische Veränderungen?
- Können Mutationen als prädiktive Marker zur Therapieentscheidung genutzt werden, um verschiedene Therapien für unterschiedliche Patienten auszuwählen?
- Gibt es derzeit schon klinische Medikamente, die gegen individuelle Mutationen eingesetzt werden können?
Bei der AML werden mittlerweile mehrere Mutationen wie FLT3 oder NPM1 routinemäßig als molekulare Marker zusätzlich zu den zytogenetischen Befunden in die Risikostratifizierung einbezogen [5], um Entscheidungen hinsichtlich Chemotherapie und Knochenmarktransplantation zu treffen.
Die momentane Risikostratifizierung bei MDS baut auf dem IPPS-R auf, der klinische, zytogenetische und histologische Parameter berücksichtigt, bislang aber noch keine Mutationen (Tab. 1) [6].
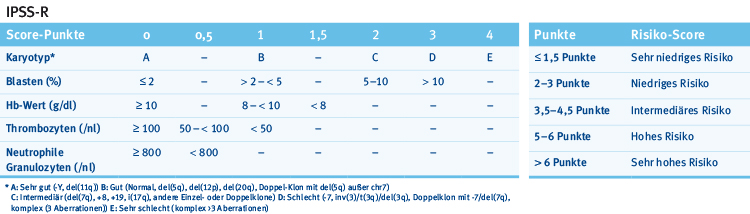 Tab. 1: IPSS-R (International Prognostic Scoring System-Revised) (nach [6]).
Tab. 1: IPSS-R (International Prognostic Scoring System-Revised) (nach [6]).Mutationen tragen zur Prognostizierung des Überlebens bei MDS-Patienten bei. Wie Abb. 1 und eine Übersicht der Onkopedia-Leitlinie zum MDS [7] zeigen, gehen die meisten Mutationen einzeln betrachtet mit einer neutralen oder ungünstigen Prognose einher (Ausnahme: SF3B1).
Die übergeordneten relevanten klinischen Fragestellungen sind aber:
- Wie beeinflussen Mutationen das Ansprechen auf hypomethylierende Substanzen (HMA) wie Azacitidin oder Decitabin?
- Haben Mutationen einen Einfluss auf das Ansprechen auf Lenalidomid bei MDS mit niedrigem Risiko mit oder ohne 5q-Deletionen (del5q)?
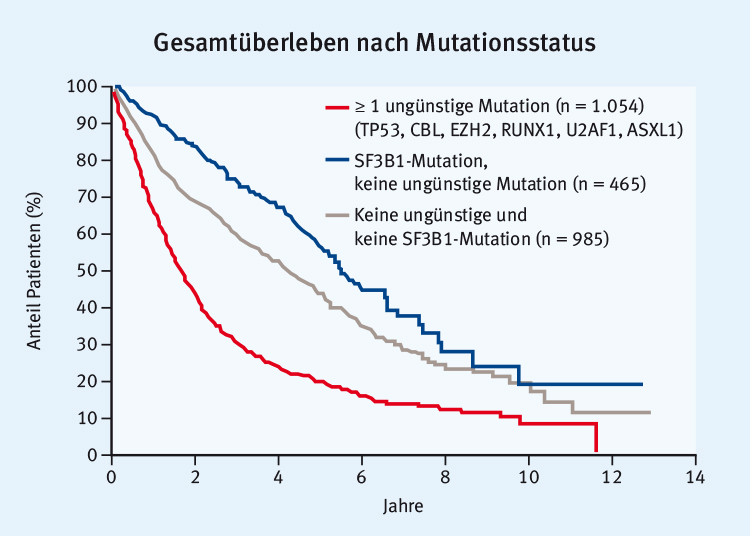 Abb. 1: Kürzeres Gesamtüberleben bei ungünstigen (adverse) Mutationen. Eine SF3B1-Mutation geht nicht mit einem kürzeren Gesamtüberleben einher (modifiziert nach [8]).
Abb. 1: Kürzeres Gesamtüberleben bei ungünstigen (adverse) Mutationen. Eine SF3B1-Mutation geht nicht mit einem kürzeren Gesamtüberleben einher (modifiziert nach [8]).Auch wenn einige Studien derzeit andeuten, dass manche Mutationen prädiktiv sein können und mit einem besseren (z. B. TET2) oder schlechteren (z. B. ASXL1) Ansprechen auf Azacitidin oder Decitabin einhergehen, ist die Überlebenswahrscheinlichkeit nicht verändert, wie exemplarisch in Abb. 2 für TET2 dargestellt. Insgesamt machen es die Konstellationen und Interaktionen von Mutationen aus, die schließlich die Risikoprognose bestimmen. Daraus ergibt sich, dass routinemäßig zwar prognostische Aussagen getroffen werden können, jedoch noch keine Therapieentscheidung aufgrund von Mutationen. Patienten sollten deshalb immer noch nach den momentanen Standards und entsprechend der klinischen Einschätzung behandelt werden. Es wird jedoch derzeit bereits empfohlen, bei Niedrig- und Intermediär-Risikopatienten auf Mutationen von TP53, ASXL1, RUNX1 und EZH2 zu testen, allerdings mehr aus prognostischen als aus therapeutisch-interventionellen Gründen. In den nächsten Jahren ist zu erwarten, dass Mutationen in die Risikostratifizierung für Therapieentscheidungen beim MDS einfließen.
 Abb. 2: Überleben mit oder ohne TET2-Mutation bei MDS unter HMA-Therapie (modifiziert nach [9]).
Abb. 2: Überleben mit oder ohne TET2-Mutation bei MDS unter HMA-Therapie (modifiziert nach [9]).Bereits jetzt kristallisiert sich ein Zusammenhang zwischen der Anzahl gleichzeitig auftretender Mutationen und einem schlechten Ansprechen und Überleben heraus. Jedoch ist es weniger die individuelle Mutation, die Prognose und Therapieansprechen beeinflusst, als vielmehr die Konstellation verschiedener Mutationen. Zum Beispiel ist das Überleben mit einer oder mehreren „ungünstigen“ Mutationen wie in ASXL1, EZH2 oder ASXL1 plus SRSF2 verkürzt, wohingegen eine isolierte SF3B1-Mutation mit einer besseren Prognose einhergeht (Abb. 1). Es gibt aber auch Daten zu einer günstigen Prognose bei mutiertem DNMT3A und nichtmutiertem (wild type) SF3B1 [8]. Diese scheinbaren Widersprüche können damit zusammenhängen, dass sich die verschiedenen Patientenkollektive und Datensätze unterscheiden und zu unterschiedlichen Assoziationen führen.
Eine weitere wichtige klinische Frage ist, ob Mutationen oder zusätzliche zytogenetische Abnormalitäten das Ansprechen auf Lenalidomid bei Patienten mit Niedrigrisiko-MDS mit oder ohne Deletion 5q (del5q) beeinflussen. Eine Zusammenfassung der vorliegenden Daten zeigt, dass Patienten mit niedrig malignem MDS und del5q auch bei (<= 2) zusätzlichen chromosomalen Abnormalitäten fast gleichwertig auf Lenalidomid ansprechen wie beim klassischen (5q-)-Syndrom. Demgegenüber sind p53-Mutationen mit einem geringeren und kürzeren Ansprechen auf Lenalidomid assoziiert. In den beiden großen Studien mit (5q-)-Syndrom erreichten 56–67% der Patienten eine Transfusionsunabhängigkeit mit einer mittleren Dauer von 83 Wochen bzw. > 2 Jahren [10, 11]. Nun haben zwei weitere größere Studien gezeigt, dass beim niedrig malignen MDS ohne Deletion 5q immer noch bis zu 27% der Patienten transfusionsunabhängig werden, im Durchschnitt mit 33 bzw. 41 Wochen jedoch kürzer [11, 33]. Daten von einer explorativen Phase-III-Studie zeigten zudem, dass bis zu 39% der Patienten auf die Kombination von Lenalidomid und Erythropoetin-stimulierenden Substanzen (ESAs) ansprachen, wenn ESAs alleine nicht mehr wirkten [12].
Zusammenfassend kann gesagt werden, dass Lenalidomid bei Patienten mit del5q und zusätzlichen chromosomalen Veränderungen eingesetzt werden kann, dass Lenalidomid beim Niedrigrisiko-MDS ohne del5q wirksam ist und dass TP53-Mutationen einen negativen Einfluss auf das Therapieergebnis haben, was jedoch eine Therapie mit Lenalidomid nicht ausschließt. Lenalidomid ist in der EU zugelassen für Niedrigrisiko-MDS mit isolierter del5q-Mutation bei Patienten, die transfusionspflichtig sind.
Zur Rolle von Mutationen bei der MDS-Diagnose und ob Mutationsanalysen routinemäßig durchgeführt werden sollen, wurde festgestellt, dass Mutationen herangezogen werden können, um eine Klonalität von Knochenmarkerkrankungen zu untermauern. Der diagnostische Wert einer solchen Analyse ist jedoch beschränkt, da bei ca. 5–22% der normalen Bevölkerung mit zunehmendem Alter (ab dem 60.–65. Lebensjahr, aber auch davor) Mutationen in den gleichen Genen (z. B. DNMT3A, TET2, ASXL1) auftreten wie beim MDS – die sogenannte „age-related clonal hematopoiesis“. Das heißt, MDS-spezifische Mutationen sind nur im klinischen Kontext diagnostisch zu verwerten, wohingegen positive MDS-spezifische zytogenetische (chromosomale) Veränderungen wie Translokationen oder Deletionen ein diagnostisches Kriterium sind.
Aber nicht jede Knochenmarkinsuffizienz stellt ein MDS dar. Es müssen unter anderem Vitaminmangel (B12-, Folat- oder Kupfermangel), HIV, Hepatitis und andere virale Erkrankungen ausgeschlossen werden, außerdem Alkoholkonsum und Autoimmunerkrankungen.
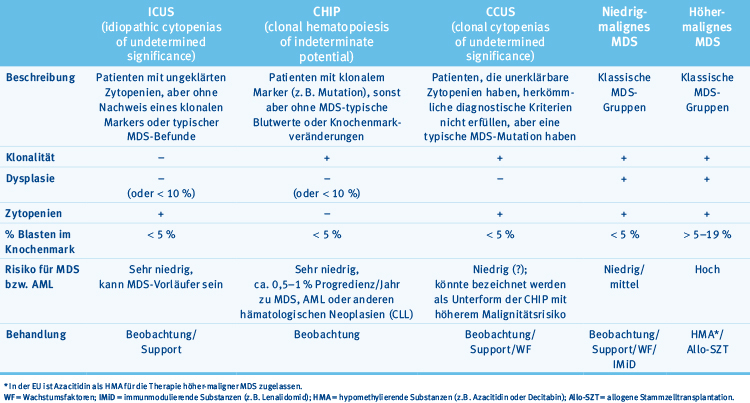
Um ungeklärte Zytopenien besser zu charakterisieren und mithilfe von Mutationsanalysen Klonalität nachzuweisen, wenn keine zytogenetischen Veränderungen nachgewiesen werden können, wurden in den letzten Jahren mehrere neue Syndrome beschrieben. Diese fallen in den Bereich der Knochenmarkinsuffizienz und können Vorstufen eines MDS sein. Sie sind vergleichbar mit dem MGUS (Monoclonal gammopathy of undetermined significance) beim multiplen Myelom oder der MBL (Monoclonal B-cell lymphocytosis) bei der CLL. Die wesentlichen Merkmale sind in Tab. 2 kurz zusammengefasst. Die wichtigsten Unterscheidungsmerkmale sind Klonalitätsmarker und das Vorliegen von Dysplasien.
Von allen Referenten wurde die neue Einordnung von MDS und AML im Rahmen der WHO-Klassifizierung hervorgehoben, die im April 2016 publiziert wurde. Auch wenn auf diese neue Klassifikation nur hingewiesen werden soll [7, 16], wurde von den Referenten zusammenfassend dargestellt, dass zelluläre Dysplasien bei der Diagnose einer MDS-assoziierten AML keinen Einfluss auf die Therapie nehmen sollten. Zudem haben therapieassoziierte MDS und AML im Vergleich zu neuauftretenden Fällen andere molekulare und zytogenetische Charakteristika, weshalb in Zukunft neue Klassifikations- und prognostische Scores definiert werden.
Fazit
- Somatische Mutationen bei MDS sind häufig. Sie sind vor allem als prognostische Faktoren zusätzlich zu den bekannten klinischen Risikofaktoren zu werten. Mutationen sollten momentan noch nicht routinemäßig als prädiktive Selektionsmarker für oder gegen bestimmte Therapien eingesetzt werden.
- Die Ausnahme von dieser Regel ist eine del5q-Mutation, die sowohl isoliert als auch zusammen mit anderen zytogenetischen Veränderungen eine hohe Ansprechrate auf Lenalidomid vorhersagt. P53-Mutationen haben einen negativen Einfluss, sollten aber nicht zum Ausschluss der Behandlung mit Lenalidomid bei del5q führen. Auch Patienten mit non-del5q-MDS können auf Lenalidomid ansprechen.
- Mutationsanalysen bei klinischem Verdacht (z. B. bei Zytopenie) und Erstdiagnose von MDS können durchgeführt werden und unter Umständen eine Klonalität bestätigen. So kann eines der Zytopeniesyndrome (ICUS, CHIP, CCUS) nachgewiesen werden. Eine Mutation allein hat aber keinen diagnostischen Wert, da ein hoher Anteil der normalen Bevölkerung ebenfalls geringe klonale Veränderungen der Blutbildung aufweist – vor allem im Alter.
- Die neue AML-WHO-Klassifikation 2016 führt zu einer verbesserten Klassifizierung. Dysplasien sollen aber nicht maßgeblich zu prognostischen oder therapeutischen Schritten herangezogen werden.
- Therapieassoziiertes MDS und AML stellt eine eigene Gruppe dar, die es noch besser zu charakterisieren gilt.
„Das Verständnis von molekularen Markern wie Mutationen wird für Diagnose, Prognose und Behandlung von MDS-Patienten zunehmend wichtig. Mutationsanalysen müssen aber immer im klinischen Kontext gesehen werden und sollten nicht zur routinemäßigen Änderung der momentan üblichen Therapien führen.“ Dr. Raoul Tibes
Session 2: Interaktive Falldiskussion zu Diagnosestellung und Prognoseeinschätzung/Prädiktion [17,18,19]
Die interaktive Falldiskussion zu Diagnosestellung und Prognoseeinschätzung war eine überaus spannende Zusammenstellung von Vorträgen, auf die im Einzelnen nicht eingegangen werden kann. Die Referenten erwähnten die neue WHO-Klassifikation 2016 für AML und MDS. Es wurde außerdem festgestellt, dass die Analyse von Mutationen vor allem bei MDS-Patienten mit niedrigem und intermediärem Risiko von Bedeutung ist. Das gilt vor allem für TP53, ASXL1, RUNX1 und EZH2, was deshalb sogar Eingang in die Onkopedia-Leitlinie zum MDS gefunden hat [7]. FACS-Analysen (Durchflusszytometrie; FACS = Fluorescence Activated Cell Sorting) können herangezogen werden, um den Phänotyp und die Anzahl der befallenen Zelllinien (uni- vs. multilineage dysplasia) näher zu charakterisieren oder in Fällen, in denen Morphologie, Zytogenetik und Mutationsanalyse kein Ergebnis zeigen. Es wurde nochmals darauf hingewiesen, dass Mutationsanalysen überwiegend einen prognostischen Wert haben. Die Abklärung und Diagnose von MDS und AML bleibt ansonsten unverändert. Bei den heterogenen therapieassoziierten MDS sind die normalen Risikoscores (IPSS-R) anwendbar, sollten aber erweitert werden.
Für die AML wurde diskutiert, dass die WHO-Überarbeitung die AML-Klassifizierung verbessert, dass der FLT3-Inhibitor Midostaurin wirksam ist, dass der CD33-Antikörper Gemtuzumab-Ozogamicin, der aktuell nicht auf dem Markt ist, evtl. in Zukunft wieder verfügbar sein könnte und dass ein „minimal residual disease“ (MRD)-Assessment bei der AML von Nutzen ist und verfolgt werden sollte. Auch wurde Wert auf eine Familienanamnese gelegt, da es eine neue WHO-Entität „familiäre AML/MDS“ gibt.
Session 3: Special Lectures: Seltene myeloische Erkrankungen
Systemische Mastozytose (SM) [20]
Bei der Mastozytose wird klinisch die indolente Form (ISM) von der fortgeschrittenen Mastozytose (ASM) mit zunehmender Organinfiltration von Mastzellen in sämtlichen Organen inklusive Knochenmark, Gastrointestinaltrakt, Leber und Herz mit erhöhten Serum-Tryptase-Spiegeln unterschieden. Häufig ist eine Eosinophilie und ein 80–90%iges pathogenetisches Auftreten der KIT D816V-Mutation mit der Mastozytose assoziiert. Bei der genetischen Charakterisierung wurden Fortschritte gemacht, häufig finden sich zusätzliche Mutationen in den typischen myeloischen Genen. So haben ca. 60% der Patienten zusätzlich zur KIT D816V-Mutation mindestens 2 weitere Mutationen. Ähnlich wie bei MDS gilt auch bei der Mastozytose: je mehr Mutationen, desto schlechter der klinische Verlauf.
Hervorgehoben wurde, dass die Mastozytose gemeinsam mit zusätzlichen aggressiven hämatologischen und myeloischen, aber auch lymphatischen Erkrankungen (SM with associated hematologic neoplams, SM-AHN) auftreten kann. Zu diesen gehören AML, MDS und myeloproliferative Neoplasien (MPN). Dabei gibt es einen breiten Überlappungsbereich, die „MDS-MPN-Overlap-Syndrome“. Diese stellen eine große Gruppe dar, die es in den kommenden Jahren besser zu charakterisieren gilt [21].
Therapeutisch zeigt Midostaurin (momentan zugelassen für die FLT3-positive AML) auch bei der ASM eine gute Ansprechrate von 60%, mit einem medianen Überleben von 28,7 Monaten [22]. In einer kleineren Studie zeigte auch Nilotinib eine Wirkung bei der ASM [23]. Bei einer sehr weit fortgeschrittenen Mastozytose (z. B. mit AML) hilft nur eine Induktionstherapie mit allogener Transplantation, aber auch diese zeigt Langzeitüberlebensraten von nur etwas über 40% und ist lediglich für jüngere und fitte Patienten geeignet.
Chronische myelomonozytäre Leukämie (CMML) [24]
Ein weiterer Vortrag sprach die diagnostischen und therapeutischen Herausforderungen bei der CMML an, die Elemente des MDS und einer myeloproliferativen Erkrankung vereint. Bei der CMML finden sich gleichzeitig Dysplasien, eine Proliferation (meistens der Leukozyten) und eine Monozytose. Auch hier gibt es einen neuen WHO-Vorschlag, die CMML in CMML 0, 1 und 2 einzuteilen. Differenzialdiagnostisch müssen eine Philadelphia-Chromosom-positive Leukämie (Ausschluss von t(9;22) und BCR-ABL) oder eine CMML mit Translokation des PDGF-A- oder B-Rezeptors ausgeschlossen werden, da letztere ausgezeichnet auf Imatinib ansprechen. Es gibt mehrere prognostische Scoringmodelle, die speziell für die CMML entwickelt wurden [25, 26]. Für Therapie und Prognose ist es relevant, ob es sich um eine eher dysplastische CMML (MDS-ähnlich mit Dysplasie und hämatopoetischer Insuffizienz) mit Leukozyten < 12.000/μl oder um eine proliferative CMML (Organo- bzw. Splenomegalie, konstitutionelle Symptome) mit Leukozyten > 12.000/μl handelt.
Die therapeutischen Optionen bei CMML sind immer noch limitiert. Azacitidin (zugelassen in der EU für CMML mit 10–29% Blasten) oder Decitabin (off-label) können eingesetzt werden. Für jüngere Patienten steht die allogene Stammzelltransplantation als Option zur Verfügung, wobei CMML-Patienten eine hohe transplantationsassoziierte Mortalität von bis zu 60% haben können. Hydroxyurea (HU) hat antiproliferative Effekte. In mehreren kleineren Studien war jedoch Azacitidin HU überlegen: z. B. zeigte sich bei 14 Azacitidin- versus 15 HU-behandelten Patienten ein Ansprechen (ORR = overall response rate) von 25% gegenüber 9% und ein medianes Überleben von 19,9 gegenüber 8,7 Monaten [27]. In einer ähnlichen Studie war das mediane Überleben mit Azacitidin 27,7 Monate und mit HU 6,2 Monate [51]. Bei bis zu 90% aller CMML-Fälle kommen Mutationen myeloischer Gene vor, jedoch mit einer anderen Inzidenz als bei AML oder MDS. In den ersten Ansätzen, molekulare Marker mit klinischen zu verbinden, zeigen Patienten mit einer ASXL1-Mutation eine ungünstige Prognose.
Akute Promyelozyten-Leukämie (APL)
Die Highlights des APL-Vortrages waren die zügige zytogenetische Abklärung und Diagnose bei Verdacht auf APL, der zügige Beginn einer Therapie mit All-Trans-Retinoinsäure (ATRA) sowie die Thematisierung der ausgeprägten Koagulopathie [28]. Die Todesrate bei den über 60-Jährigen ist mit ca. 29% immer noch sehr hoch, meist bedingt durch Blutungskomplikationen. Die Kombination von Arsentrioxid (ATO) und ATRA ist hocheffektiv und nur 1,3% der Patienten haben ein Rezidiv. Diese Kombination wird jetzt auch bei den APL-Patienten mit höherem Risiko eingesetzt und kann mindestens 50% der Patienten mit Rezidiv nach ATRA- und Chemotherapie-Induktion heilen.
Fazit
- Die fortgeschrittene systemische Mastozytose (ASM) ist ein seltenes, aber hoch akutes Krankheitsbild und kann zusammen mit anderen hämatologischen Neoplasien auftreten (SM-AHN). Genetische Mutationen sind häufig und gehen mit einer negativen Prognose einher. Midostaurin und Nilotinib können wirksam sein.
- Die CMML ist ein sogenanntes Overlap-Syndrom. Klinisch wichtig in Bezug auf Präsentation und Therapien ist die Unterscheidung zwischen der dysplastischen (MDS-typisch) und der myeloproliferativen CMML. Azacitidin kann angewendet werden, mit moderaten Ansprechraten. Genetische Mutationen spielen auch bei der CMML eine Rolle und werden zunehmend in Therapieentscheidungen integriert.
- APL ist eine seltene Erkrankung, aber heilbar. Die frühe Sterblichkeit ist hoch, überwiegend bedingt durch eine massive Koagulopathie mit Blutungen. Rasche Diagnose, schneller Start einer ATRA-Therapie, eine enge klinische Betreuung mit Korrektur der Koagulopathie und Plättchentransfusion sind essenziell. Die Kombination von ATRA und ATO hat bei niedrigem und intermediärem Risiko sehr hohe Heilungsraten (> 90–95%) und wird nun auch bei Hochrisiko-APL klinisch getestet (APOLLO-Studie).
„Die seltenen myeloproliferativen/myelodysplastischen Erkrankungen stellen eine Herausforderung dar und sind in ihrer Gesamtheit doch recht häufig. Über die nächsten Jahre werden auch diese zunehmend genetisch profiliert, was hoffentlich zu weiteren Therapiemöglichkeiten führen wird. Momentan werden Therapiestrategien sowohl aus dem MDS als auch dem MPN-Bereich bei der CMML angewendet.“ Dr. Raoul Tibes
Session 4: Therapie des Niedrigrisiko-MDS [29,30,31,32]
Zunächst wurde die Pathophysiologie des (5q-)-Syndroms diskutiert und darauf hingewiesen, dass vor allem p53-Mutationen die Prognose des Niedrigrisiko-MDS negativ beeinflussen. Es wurde betont, dass Patienten auf Lenalidomid immer noch gut ansprechen, wenn außer del5q noch bis zu zwei weitere zytogenetische Aberrationen vorliegen. Eine vorgestellte Studie zeigte eine Transfusionsunabhängigkeit von 27% bzw. 18% nach 2 bzw. 6 Monaten bei non-del5q-Patienten. Je niedriger der Erythropoetinspiegel war (EPO < 100 U/l: 42,5% Ansprechen), desto besser war das Ansprechen [33]. Neben einem niedrigeren EPO-Spiegel (< 500 U/l, noch besser < 100 U/l) verbessert den Therapierfolg mit ESA (erythropoetin stimulating agents) ein kurzes Intervall (< 6 Monate) zwischen Diagnosestellung und Beginn der ESA-Therapie, ein höherer Hb (> 9 g/dl) und Vorliegen des MDS-Subtyps RCMD-RS. Dies bestätigt Ergebnisse aus der MDS-002-Studie, die sehr ähnliche Ansprechraten hatte [11].
Daten aus einer weiteren Studie zeigten, dass Erythropoetin und Lenalidomid zusammen eine Ansprechrate von rund 39% haben [12]. Bei einem EPO-Level von > 500 U/l kann Azacitidin zum Einsatz kommen und wirkt in ca. 20%. Hervorgehoben wurde auch das neue Studienmedikament Luspatercept (ACE536), das den TGF-Beta-Signalweg moduliert. In der sogenannten PACE-Trial kam es bei 40–54% der Patienten zur Transfusionsunabhängigkeit, unabhängig davon, ob Patienten vorher ein ESA-Medikament erhalten hatten oder nicht [52]. Bei Patienten mit Ringsideroblasten und/oder SF3B1-Mutation lag die Ansprechrate sogar bei 67–72%, und dies bei relativ geringen Nebenwirkungen. Hämatologische Verbesserungen der Erythropoese (HI-E) wurden bei 20–80% der Patienten in Abhängigkeit vom EPO-Spiegel beobachtet. Mehrere Studien mit Luspatercept sind aktuell dabei, Patienten mit Niedrigrisiko-MDS und Transfusionsbedarf zu rekrutieren.
In einem weiteren Vortrag wurde ein Blutungsrisiko von 10–12% bei MDS-Patienten mit Thrombozytopenie beschrieben. Die Blutungsneigung wird bei diesen Patienten nicht nur durch eine verminderte Thrombozytenzahl, sondern auch durch eine schlechtere Funktionalität der Plättchen verursacht. Thrombopoetin (TPO)-stimulierende Medikamente wie Romiplostim oder Eltrombopag können zu einer Verbesserung der Thrombozytopenie führen und haben auch einen antiproliferativen Effekt (es gab Remissionen als single-agent-Therapie bei der AML). Ob diese Medikamente bei MDS-Patienten wirklich nützlich sein werden, ist noch nicht geklärt. Nach anfänglichen Befürchtungen einer höheren AML-Inzidenz bei den MDS-Studien konnte dies im längeren Verlauf nicht bestätigt werden. Klinische Studien dauern an und es soll betont werden, dass weder Romiplostim noch Eltrombopag für MDS zugelassen sind. Bei der schweren aplastischen Anämie ist Eltrombopag hingegen zugelassen, mit Ansprechraten von bis zu 44% [34, 35].
Auch zum Thema der Eisenchelation gab es einen ausgezeichneten Vortrag. Im Gegensatz zu Thalassämien sind schwere kardiale Eisenüberladungen beim MDS selten. Eisenchelation senkt nicht die Anzahl der Infektionen, wie man aufgrund der Pathophysiologie annahm. Es gibt keine prospektiven Studien, die einen Überlebensvorteil von Eisenchelation beim MDS zeigen.
Fazit
- Je geringer der EPO-Spiegel, je höher das Hämoglobin (> 9 g/dl), je kürzer die Zeit zwischen Transfusionsabhängigkeit und Start einer ESA-Therapie (ideal < 6 Monate) und je weniger Transfusionen, desto höher ist die Ansprechrate auf ESAs. Patienten sollten daher eher früh behandelt werden.
- Luspatercept ist ein Medikament, das in der klinischen Entwicklung hohe Raten von Transfusionsunabhängigkeit bei recht geringen Nebenwirkungen gezeigt hat. Die klinische Entwicklung läuft.
- Die Rolle der TPO-Agonisten wie Eltrombopag bei MDS ist derzeit noch offen.
- Eisenchelation beim MDS ist möglich, prospektive und randomisierte Studien zum Überleben fehlen aber.
„Die größte Neuerung beim Niedrigrisiko-MDS ist sicherlich Luspatercept. Lenalidomid ist wirksam und kann eingesetzt werden, auch bei Patienten mit non-del5q-MDS.“ Dr. Raoul Tibes
Session 5: Therapie des Hochrisiko-MDS [36,37,38]
Auf diesem Gebiet hat sich momentan wahrscheinlich am wenigsten verändert. Zunächst wurde hervorgehoben, dass es momentan noch keine verlässlichen prädiktiven molekularen Marker für das Ansprechen auf HMAs gibt, diese aber benötigt werden. Das Ansprechen auf Azacitidin oder Decitabin ist relativ unabhängig von negativen prognostischen zytogenetischen Markern oder Mutationen, dem Alter (sogar bei > 75-Jährigen) oder dem Grad des Ansprechens. Therapiert werden muss für mindestens 6 Monate, bevor ein Therapieversagen festgestellt werden kann. Denn von den insgesamt ansprechenden Patienten zeigen nur 20% der Patienten bereits nach 2 Zyklen ein Ansprechen. Im Falle des Ansprechens sollte die Therapie auf jeden Fall weitergeführt werden. Der Zusatz von Lenalidomid oder Vorinostat (ein HDAC-Inhibitor) bringt keinen Vorteil gegenüber Azacitidin allein, wie in einer randomisierten Studie demonstriert wurde [39]. Bei einem Rezidiv nach allogener Transplantation ist Azacitidin allein sogar besser als eine Kombination mit Chemotherapie. Bei einem Verlust des Ansprechens auf eine HMA-Therapie kann ein Wechsel zu dem alternativen HMA durchgeführt werden, wobei die Ansprechraten dann sehr moderat und eher kurz sind (ca. 3–6 Monate). Eine Teilnahme an klinischen Studien mit einem der vielen neuen Medikamente ist daher bei HMA-Versagen eher zu empfehlen. In der EU ist Azacitidin für die Therapie nicht transplantierbarer Patienten mit Hochrisiko-MDS zugelassen. Eine allogene Stammzelltransplantation bietet die einzig mögliche Heilung, vor allem bei jüngeren Patienten.
Fazit
- Es gibt momentan keine verlässlichen prädiktiven Marker, die ein Ansprechen auf Azacitidin oder Decitabin vorhersagen können.
- Das Erreichen einer kompletten Remission ist nicht notwendigerweise für einen Überlebensvorteil unter Azacitidin erforderlich. Bei Ansprechen auf Azacitidin sollte definitiv therapiert werden.
- Bislang hat der Zusatz von anderen Medikamenten zu Azacitidin oder Decitabin keinen Vorteil gezeigt.
- Eine allogene Transplantation (falls geplant) sollte während des Maximums des HMA-Ansprechens erfolgen.
“Leider hat sich bei der Hochrisiko-MDS in den letzten Jahren nicht viel getan. Mutationen beeinflussen auch hier die Prognose, haben aber noch nicht entscheidend zur Therapieveränderung geführt. Es gibt jedoch mehrere rationale Kombinationen mit Azacitidin oder Decitabin [40, 41], die klinisch vielversprechend sein können. Bei HMA-Versagen, entweder a priori oder nach initialem Ansprechen, sind die Therapiemöglichkeiten und das Überleben äußerst begrenzt. Diese Patienten stellen eine große Herausforderung dar.“ Dr. Raoul Tibes
Session 6: Therapie des älteren AML-Patienten [42,43,44,45]
Ein besonderes klinisches Problem stellt die Gruppe der älteren AML-Patienten dar (ab > 65 Jahre, auf jeden Fall > 75 Jahre) – vor allem, wenn sie aufgrund ihres Allgemeinzustandes oder wegen Komorbiditäten keine Kandidaten für eine Induktionstherapie sind. In diesen Fällen kommt Azacitidin oder Decitabin mit Ansprechraten von 45–55% zum Einsatz, wobei selten eine komplette Remission beobachtet werden kann. Die Überlebenszeit ist mit 6–13 Monaten je nach zytogenetischer Gruppe immer noch kurz. Auch niedrig dosiertes subkutanes Cytarabin kann in 20–30% der Fälle wirksam sein. Eine Ausnahme stellen Patienten mit Core binding leukemias dar (Translokationen t(8;21) und inv(16) bzw. t(16;16)), die von einer intensiven Chemotherapie auch im Alter profitieren können [46]. In Bezug auf neue Induktionstherapien gibt es keine großen Entwicklungen. Der Zusatz von Azacitidin zu einer konventionellen Chemotherapie zeigt eine negative Tendenz hinsichtlich des Überlebens, wenngleich nicht statistisch signifikant [47].
Bei der allogenen Transplantation hat sich gezeigt, dass die reduzierte Konditionierung unter Umständen weniger effektiv ist und bis zu 50% der AML- und 37% der MDS-Patienten ein Rezidiv erleiden, wohingegen bei einer voll myeloablativen Konditionierung nur 16,5% der AML und 3,7% der MDS rezidivieren. Daher wird diskutiert, ob die myeloablative Konditionierung wieder bevorzugt eingesetzt werden soll. In Bezug auf MRD zeigt sich, dass eine Reduktion von NPM1 auf weniger als 10% eine günstige Langzeit-Überlebensprognose darstellt.
Hinsichtlich neuer Therapien bei einem AML-Rezidiv wurden Daten zu dem IDH2-Inhibitor AG-221 vorgestellt, auf den 41% der AML-Patienten eine komplette oder partielle Remission zeigten, die im Durchschnitt mehr als 15 Monate dauerte [48]. Die Ansprechrate auf AG-120 bei Patienten mit IDH1-mutierter AML lag bei 31% mit einem medianen Ansprechen von 11 Monaten [49]. Mit diesen herausragenden Ergebnissen sollten AML-Patienten im Rezidiv auf IDH1- oder IDH2-Mutationen getestet werden. Auch der CD33-Antikörper Gemtuzumab-Ozogamicin kann allein oder in Kombination mit Chemotherapie beim AML-Rezidiv wirksam sein, wie die Referentin zusammenfasste. In einer kleinen Studie mit rezidivierten AML- und MDS-Patienten hat der CTLA-4-Immunantikörper Ipilimumab bei 42% der Patienten gewirkt, darunter auch bei Patienten mit Leukemia cutis und myeloischem Sarkom [50]. Letztendlich können auch FLT3-Inhibitoren bei FLT3-Mutationen wirken und sollten im Rahmen einer klinischen Studie berücksichtigt werden.
Fazit
- Azacitidin und Decitabin (allein oder in Kombinationsstudien) sind für unfitte ältere AML-Patienten Standard, unabhängig von negativen molekularen Risikofaktoren. Niedrig dosiertes Cytarabin ist eine Alternative. Die Ausnahme stellen AML-Patienten mit t(8;22) und t(16;16), inv(16) dar, die mit klassischer Chemotherapie behandelt werden sollten.
- Medikamente in der klinischen Entwicklung wie Inhibitoren von FLT3, IDH1 (AG-120) oder IDH-2 (AG-221) können sehr wirksam sein, wobei die Ansprechdauer wichtig ist.
- Immuncheckpoint-Therapien (z. B. PD-1, PD-L1, CTLA-4) halten auch bei der AML-Therapie Einzug.
- Die allogene Transplantation ist eine therapeutische Option, bei der zunehmend wieder myeloablative Konditionierungstherapien gewählt werden, da die Rezidivrate nach einer reduzierten Konditionierungstherapie recht hoch ist.
„Auch wenn HMAs den Standard für ältere AML-Patienten darstellen, sind das Gesamtansprechen und das Überleben noch suboptimal. Deshalb müssen neue rationale Therapien entwickelt werden. Zu den molekularen Therapiezielen, für die Medikamente in der klinischen Entwicklung sind, gehören Inhibitoren von FLT3, IDH1 und IDH2.“ Dr. Raoul Tibes
Quellen
- Bejar R. Pathophysiologie bei MDS und AML. MDS-Forum, April 2016, München.
- Haferlach T. AML mit myelodysplasia-related changes: Eine relevante Entität? MDS-Forum, April 2016, München.
- Kündgen A. Therapieassoziierte MDS/AML: Müssen wir umdenken? MDS-Forum, April 2016, München.
- Bejar R, Stevenson K, Abdel-Wahab O et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011; 364: 2496-2506.
- Döhner H, Estey EH, Amadori S et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115: 453-474.
- Greenberg PL, Tuechler H, Schanz J et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012; 120: 2454-2465.
- Onkopedia Leitlinien. Myelodysplastische Syndrome (MDS); verfügbar unter https://www.onkopedia.com/de/onkopedia/guidelines/myelodysplastische-syndrome-mds; abgerufen am 02.05.2016.
- Bejar R, Papaemmanuil E, Haferlach T et al. Somatic Mutations in MDS Patients Are Associated with Clinical Features and Predict Prognosis Independent of the IPSS-R: Analysis of Combined Datasets from the International Working Group for Prognosis in MDS-Molecular Committee. ASH 2015, Orlando, abstract 907.
- Bejar R, Lord A, Stevenson K et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014; 124: 2705-2712.
- Fenaux P, Giagounidis A, Selleslag D et al. A randomized phase 3 study of lenalidomide versus placebo in RBC transfusion-dependent patients with Low-/Intermediate-1-risk myelodysplastic syndromes with del5q. Blood 2011; 118: 3765-3776.
- Raza A, Reeves JA, Feldman EJ et al. Phase 2 study of lenalidomide in transfusion-dependent, low-risk, and intermediate-1 risk myelodysplastic syndromes with karyotypes other than deletion 5q. Blood 2008; 111: 86-93.
- Toma A, Kosmider O, Chevret S et al. Lenalidomide with or without erythropoietin in transfusion-dependent erythropoiesis-stimulating agent-refractory lower-risk MDS without 5q deletion. Leukemia 2016; 30: 897-905.
- Valent P, Bain BJ, Bennett JM et al. Idiopathic cytopenia of undetermined significance (ICUS) and idiopathic dysplasia of uncertain significance (IDUS), and their distinction from low risk MDS. Leuk Res 2012; 36: 1-5.
- Steensma DP, Bejar R, Jaiswal S et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood 2015; 126: 9-16.
- Kwok B, Hall JM, Witte JS et al. MDS-associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood 2015; 126: 2355-2361.
- Arber DA, Orazi A, Hasserjian R et al. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood 2016.
- Germing U, Haase D, Müller-Thomas C et al. 4 Fälle mit interaktiver Diskussion im Panel. MDS-Forum, April 2016, München.
- Haase D. Notwendige Diagnostik bei MDS in 2016. MDS-Forum, April 2016, München.
- Schlenk R. Notwendige Diagnostik bei AML in 2016. MDS-Forum, April 2016, München.
- Reiter A. Mastozytose bei MDS und assoziierten Erkrankungen. MDS-Forum, April 2016, München.
- Mughal TI, Cross NC, Padron E et al. An International MDS/MPN Working Group's perspective and recommendations on molecular pathogenesis, diagnosis and clinical characterization of myelodysplastic/myeloproliferative neoplasms. Haematologica 2015; 100: 1117-1130.
- Gotlib J, Kluin-Nelemans HC, George TI et al. Midostaurin (PKC412) Demonstrates a High Rate of Durable Responses in Patients with Advanced Systemic Mastocytosis: Results from the Fully Accrued Global Phase 2 CPKC412D2201 Trial. Blood 2014; 124: Abstract 636.
- Hochhaus A, Baccarani M, Giles FJ et al. Nilotinib in patients with systemic mastocytosis: analysis of the phase 2, open-label, single-arm nilotinib registration study. J Cancer Res Clin Oncol 2015; 141: 2047-2060.
- Germing U, Pleyer L. CMML: Eine diagnostische und therapeutische Herausforderung. MDS-Forum, April 2016, München.
- Onida F, Kantarjian HM, Smith TL et al. Prognostic factors and scoring systems in chronic myelomonocytic leukemia: a retrospective analysis of 213 patients. Blood 2002; 99: 840-849.
- Such E, Germing U, Malcovati L et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood 2013; 121: 3005-3015.
- Dinmohamed A, Visser O, Posthuma W et al. Clinical features, treatment decisions and treatment outcomes of patients with chronic myelomonocytic leukemia (CMML) in the real-world: results from the dutch population-based pharos MDS registry. Presented at E-poster, EHA 2015, Vienna, abstract E1214.
- Lengfelder E. APL: “State of the art”. MDS-Forum, April 2016, München.
- Giagounidis A. Lenalidomid bei Niedrigrisiko-MDS: Ändern molekulare Marker unser Vorgehen? MDS-Forum, April 2016, München.
- Götze K. ESAs und Alternativen: Konkurrenz für EPO? MDS-Forum, April 2016, München.
- Platzbecker U. Thrombozytopenie: Renaissance der TPO-Agonisten? MDS-Forum, April 2016, München.
- Wermke M. Eisenüberladung: ALLIVE and kicking durch Chelation? MDS-Forum, April 2016, München.
- Santini V, Almeida A, Giagounidis A et al. Efficacy and Safety of Lenalidomide (LEN) Versus Placebo (PBO) in RBC-Transfusion Dependent (TD) Patients (Pts) with IPSS Low/Intermediate (Int-1)-Risk Myelodysplastic Syndromes (MDS) without Del(5q) and Unresponsive or Refractory to Erythropoiesis-Stimulating Agents (ESAs): Results from a Randomized Phase 3 Study (CC-5013-MDS-005). Blood 2014; 124: Abstract 409.
- Olnes MJ, Scheinberg P, Calvo KR et al. Eltrombopag and improved hematopoiesis in refractory aplastic anemia. N Engl J Med 2012; 367: 11-19.
- Erratum zu Olnes MJ 2012: Eltrombopag and Improved Hematopoiesis in Refractory Aplastic Anemia. New England Journal of Medicine 2012; 367: 284-284.
- Bug G. First-Line-Therapie des Hochrisiko-MDS: HMA und darüber hinaus? MDS-Forum, April 2016, München.
- Hofmann W-K. Prädiktive Marker für das Ansprechen auf HMA: Ein frommer Wunsch? MDS-Forum, April 2016, München.
- Schroeder T. Optionen nach HMA-Versagen: Licht am Horizont? MDS-Forum, April 2016, München.
- Sekeres MA, Othus M, List AF et al. A Randomized Phase II Study of Azacitidine Combined with Lenalidomide or with Vorinostat Vs. Azacitidine Monotherapy in Higher-Risk Myelodysplastic Syndromes (MDS) and Chronic Myelomonocytic Leukemia (CMML): North American Intergroup Study SWOG S1117. Blood 2014; 124: Abstract LBA-5.
- Bogenberger JM, Kornblau SM, Pierceall WE et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia 2014; 28: 1657-1665.
- Issa JP, Roboz G, Rizzieri D et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol 2015; 16: 1099-1110.
- Bornhäuser M. Allogene Stammzelltransplantation für MDS/AML-Patienten > 60 Jahre: one size fits all? MDS-Forum, April 2016, München.
- Döhner H. Epigenetische Therapie: Für alle „elderly“ AML-Patienten? MDS-Forum, April 2016, München.
- Müller-Tidow C. Intensive Chemotherapie: Endlich mehr als nur „7 + 3“? MDS-Forum, April 2016, München.
- Thol F. Rezidivierte AML: Optionen jenseits der Transplantation? MDS-Forum, April 2016, München.
- Paschka P, Schlenk RF, Weber D et al. Dasatinib (DAS) in combination with chemotherapy and as maintenance in core-binding factor (CBF) acute myeloid leukemia (AML): A phase Ib/IIa study of the german-austrian AML study group (AMLSG). Presented at Oral Presentation, EHA 2015, Vienna, abstract S515.
- Müller-Tidow C, Tschanter P, Rollig C et al. Azacitidine in combination with intensive induction chemotherapy in older patients with acute myeloid leukemia: The AML-AZA trial of the study alliance leukemia. Leukemia 2016; 30: 555-561.
- Stein EM, DiNardo C, Altman JK et al. Safety and Efficacy of AG-221, a Potent Inhibitor of Mutant IDH2 That Promotes Differentiation of Myeloid Cells in Patients with Advanced Hematologic Malignancies: Results of a Phase 1/2 Trial. ASH 2015, Orlando, abstract 323.
- DiNardo C, de Botton S, Pollyea DA et al. Molecular Profiling and Relationship with Clinical Response in Patients with IDH1 Mutation-Positive Hematologic Malignancies Receiving AG-120, a First-in-Class Potent Inhibitor of Mutant IDH1, in Addition to Data from the Completed Dose Escalation Portion of the Phase 1 Study. ASH 2015, Orlando, abstract 1306.
- Davids MS, Kim HT, Costello CL et al. A Multicenter Phase I/Ib Study of Ipilimumab for Relapsed Hematologic Malignancies after Allogeneic Hematopoietic Stem Cell Transplantation. ASH 2015, Orlando, abstract 860.
- Pleyer L, Germing U, Sperr WR et al. Azacitidine in CMML: matched-pair analyses of daily-life patients reveal modest effects on clinical course and survival. Leuk Res 2014; 38: 475-483.
- Giagounidis A, Platzbecker U, Germing U et al. Luspatercept Treatment Leads to Long Term Increases in Hemoglobin and Reductions in Transfusion Burden in Patients with Low or Intermediate-1 Risk Myelodysplastic Syndromes (MDS): Preliminary Results from the Phase 2 PACE-MDS Extension Study. ASH 2015, Orlando, abstract 92.
- Bildnachweis: „The Frauenkirche is a church in the Bavarian city of Munich”: © Jörg Hackemann/Fotolia; "Munich cuty center skyline": © R.Babakin/Fotolia
Session 6: Therapie des älteren AML-Patienten [42-45]
Ein besonderes klinisches Problem stellt die Gruppe der älteren AML-Patienten dar (ab > 65 Jahre, auf jeden Fall > 75 Jahre) – vor allem, wenn sie aufgrund ihres Allgemeinzustandes oder wegen Komorbiditäten keine Kandidaten für eine Induktionstherapie sind. In diesen Fällen kommt Azacitidin oder Decitabin mit Ansprechraten von 45–55% zum Einsatz, wobei selten eine komplette Remission beobachtet werden kann. Die Überlebenszeit ist mit 6–13 Monaten je nach zytogenetischer Gruppe immer noch kurz. Auch niedrig dosiertes subkutanes Cytarabin kann in 20–30% der Fälle wirksam sein. Eine Ausnahme stellen Patienten mit Core binding leukemias dar (Translokationen t(8;21) und inv(16) bzw. t(16;16)), die von einer intensiven Chemotherapie auch im Alter profitieren können [46]. In Bezug auf neue Induktionstherapien gibt es keine großen Entwicklungen. Der Zusatz von Azacitidin zu einer konventionellen Chemotherapie zeigt eine negative Tendenz hinsichtlich des Überlebens, wenngleich nicht statistisch signifikant [47].
Bei der allogenen Transplantation hat sich gezeigt, dass die reduzierte Konditionierung unter Umständen weniger effektiv ist und bis zu 50% der AML- und 37% der MDS-Patienten ein Rezidiv erleiden, wohingegen bei einer voll myeloablativen Konditionierung nur 16,5% der AML und 3,7% der MDS rezidivieren. Daher wird diskutiert, ob die myeloablative Konditionierung wieder bevorzugt eingesetzt werden soll. In Bezug auf MRD zeigt sich, dass eine Reduktion von NPM1 auf weniger als 10% eine günstige Langzeit-Überlebensprognose darstellt.
Hinsichtlich neuer Therapien bei einem AML-Rezidiv wurden Daten zu dem IDH2-Inhibitor AG-221 vorgestellt, auf den 41% der AML Patienten eine komplette oder partielle Remission zeigten, die im Durchschnitt mehr als 15 Monate dauerte [48]. Die Ansprechrate auf AG-120 bei Patienten mit IDH1-mutierter AML lag bei 31% mit einem medianen Ansprechen von 11 Monaten [49]. Mit diesen herausragenden Ergebnissen sollten AML-Patienten im Rezidiv auf IDH1- oder IDH2-Mutationen getestet werden. Auch der CD33-Antikörper Gemtuzumab-Ozogamicin kann allein oder in Kombination mit Chemotherapie beim AML-Rezidiv wirksam sein, wie die Referentin zusammenfasste. In einer kleinen Studie mit rezidivierten AML- und MDS-Patienten hat der CTLA-4-Immunantikörper Ipilimumab bei 42% der Patienten gewirkt, darunter auch bei Patienten mit Leukemia Cutis und Myeloischem Sarkom [50]. Letztendlich können auch FLT3-Inhibitoren bei FLT3-Mutationen wirken und sollten im Rahmen einer klinischen Studie berücksichtigt werden.
Fazit
- Azacitidin und Decitabin (allein oder in Kombinationsstudien) sind für unfitte ältere AML-Patienten Standard, unabhängig von negativen molekularen Risikofaktoren. Niedrig dosiertes Cytarabin ist eine Alternative. Die Ausnahme stellen AML-Patienten mit t(8;22) und t(16;16), inv(16) dar, die mit klassischer Chemotherapie behandelt werden sollten.
- Medikamente in der klinischen Entwicklung wie Inhibitoren von FLT3, IDH1 (AG-120) oder IDH-2 (AG-221) können sehr wirksam sein, wobei die Ansprechdauer wichtig ist.
- Immuncheckpoint-Therapien (z. B. PD-1, PD-L1, CTLA-4) halten auch bei der AML-Therapie Einzug.
- Die allogene Transplantation ist eine therapeutische Option, bei der zunehmend wieder myeloablative Konditionierungstherapien gewählt werden, da die Rezidivrate nach einer reduzierten Konditionierungstherapie recht hoch ist.
„Auch wenn HMAs der Standard für ältere AML-Patienten darstellen, sind das Gesamtansprechen und das Überleben noch suboptimal. Deshalb müssen neue rationale Therapien entwickelt werden. Zu den molekularen Therapiezielen, für die Medikamente in der klinischen Entwicklung sind, gehören Inhibitoren von FLT3, IDH1 und IDH2.“ Dr. Raoul Tibes